Step 3: MagIC-cryo-EM Beads Quality Assessment
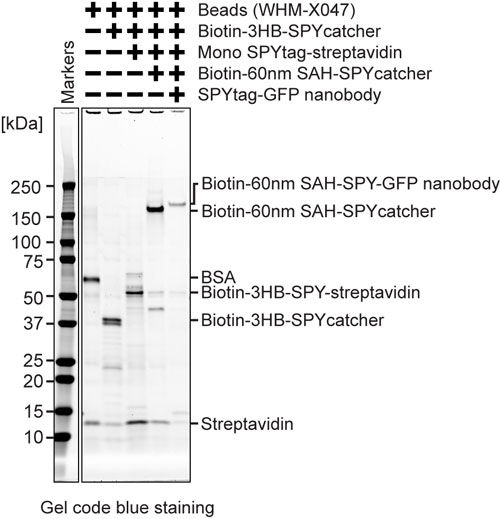
MagIC-cryo-EM beads quality assessment (SDS-PAGE)
SDS-PAGE of 2~5 fmol beads can visualize the protein bands. We highly recommend running a SDS-PAGE of each step of the beads, including the streptavidin beads, before assembling the proteins (SDS-PAGE should show the bands of BSA and streptavidin).
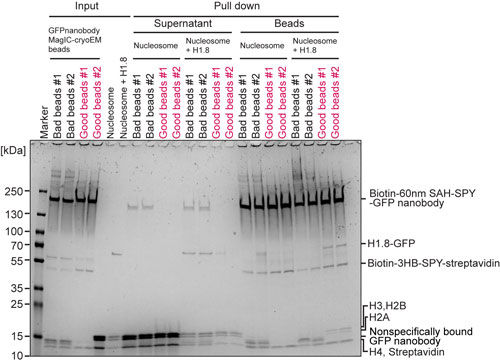
MagIC-cryo-EM beads quality assessment (Pull-down assay)
Confirming the pulldown of GFP-tagged proteins is useful for assessing the quality of the beads before using them for the cryo-EM sample, as it ensures that the sample won’t be wasted. Below is the quality check experiment done in our lab. ‘GFP-H1.8-nucleosome’ and ‘nucleosome’ can be replaced with both your positive and negative control samples. (Please contact me if you want to learn what caused the “bad beads” in the gel.)
- Mix GFP-nanobody MagIC-cryo-EM beads with 5 pmol nucleosome or 2 pmol GFP-H1.8-nucleosome in 50 µL solution
- Place on ice 2 hours
- Spin tubes at 16000g, 20min, 4ºC.
- Transfer tubes onto the handmade magnetic rack and remove the supernatant
- Resuspend the beads with 100 µL of wash buffer
- Spin the tubes at 16000g, 20min, 4ºC.
- Transfer the tubes onto the handmade magnetic rack and remove the supernatant
- Resuspend the beads with SDS sample buffer and run SDS-PAGE
MagIC-cryo-EM*
This is the test experiment to.
* For the nucleosome, 1000~2000 nucleosomes/bead is an ideal ratio. This may vary depending on your target.
Step 1: To remove too small beads with the same buffer for the sample
- Mix 10 µL of 3HB-60nm-SAH-GFPe beads [1 fmol], 100 µL 17% sucrose gradient buffer with 0.01% Tween20
- Spin 16000g 20 min 4ºC, collect the tube on the handmade magnetic rack
- Remove supernatant from the tube on the handmade magnetic rack
Step 2: to remove aggregated protein
- Transfer the sample (a fraction from a sucrose gradient containing 1~2 pmol* target complex) to a new tube (* For the nucleosome, 1000~2000 nucleosomes/bead is an ideal ratio. This may vary depending on your target.)
- Add Tween20 to 0.01%
- Spin at 16000g 20 min 4ºC
- Take supernatant
Step 3: Incubation
- Add the supernatant of Step 1 to the washed beads of Step 2
- Incubate (Leave 15 hrs in the cold room) overnight
Step 4: Wash
For the purified protein, this step may not be needed.
- Spin at 16000g 20 min 4ºC
- Remove supernatant
- Add 200 µL of EM cryoprotectant buffer with 0.01% Tween
- Pipette well
- Spin at 16000g 20 min 4ºC
- Remove supernatant
- Add 200 µL of EM cryoprotectant buffer with 0.01% Tween
- Pipette well
- Spin at 16000g 20 min 4ºC
- Remove supernatant
- Add 200 µL of EM cryoprotectant buffer with 0.01% Tween
- Pipette well
- Spin at 16000g 20 min 4ºC
- Remove supernatant
- Add 80 µL of EM cryoprotectant buffer with 0.001% Tween
- Pipette well
- Freeze grids (incubate 5 min on magnets)
Step 5: Grid freezing
- Plasma clean graphene grid (O₂ + H₂, 10 sec)
- Pick one grid with non-magnetic tweezers and set it in a magnetic humidity chamber
- Apply 4 µL of sample
- Incubate 5 min
- Freeze grid with Vitrobot
- (Skip sample application, 2-second blotting time at room temperature under 100% humidity)
© 2025 Fred Hutchinson Cancer Center, a 501(c)(3) nonprofit organization.