Research
Signal Transduction and Cancer
Animals, such as us, are composed of many cells that cooperate to benefit the organism as a whole. Cells cooperate by exchanging signals that specify when they divide, when to die, when and where to move, and which specialized functions to adopt. The signals come in the form of molecules that diffuse between cells as well as contact signals that are transmitted between cells that touch each other. Many types of cells also secrete molecules that form a matrix. The cells then adhere to this matrix and the matrix signals back to the cells through the adhesion sites. Together, these various types of signals coordinate cellular activities, so that a fertilized egg can develop into an entire animal and so that an adult animal can maintain its internal state despite changes in its surroundings. Defects in cell signaling can lead to run-away cell growth, division and migration, resulting in cancer.
Signals are received by the cell through receptors on the cell surface, transduced across the membrane, and relayed by a series of chemical reactions (a signaling "pathway") to regulate cell behavior. Many signaling pathways involve protein kinases - enzymes that transfer phosphate onto other proteins, thereby altering their properties. Tyrosine kinases, which transfer phosphate onto tyrosine residues in other proteins, are particularly important, in that many of them are primary regulators of cell proliferation and migration. If they are de-regulated by mutations, tyrosine kinases can drive cancer by continuously providing signals that stimulate cell division and movement.
Previous research
In the past, the Cooper lab research has revealed different aspects of tyrosine kinase signaling. For example, we found that the cell surface receptor for platelet-derived growth factor binds other cell proteins when it is activated by phosphorylation on a specific tyrosine residue. Recruitment of these proteins to the membrane stimulates the next steps in signaling pathways that regulate cell proliferation. We also made an important connection between a small GTP hydrolysis enzyme (Ras) and a protein kinase (Raf), both of which are important in human cancers. We helped elucidate the mechanism that regulates the activity of Src, an enzyme that is mutated in a virus that causes cancer in chickens and contributes to cancer in humans. Our chance discovery of Dab1, a Src substrate (a protein phosphorylated by Src) that is expressed in the brain, led us to several years investigating how immature nerve cells (neurons) migrate during brain development.
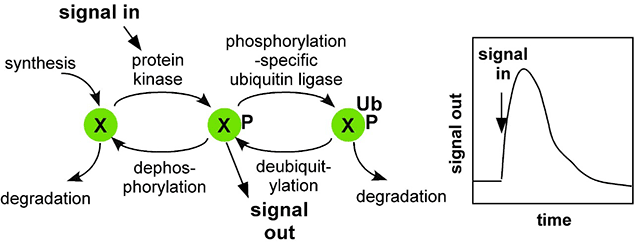
Our discovery of Dab1 came at a time when neuron migrations were known to be regulated by an external signal (Reelin). Our studies showed that Reelin stimulates Src to phosphorylate Dab1 inside migrating neurons. Dab1 phosphorylation then stimulates neuron movement by various mechanisms, including upregulating a cell-cell adhesion molecule, N-cadherin, on the cell surface. We also found that Reelin stimulates Dab1 degradation by an interesting mechanism. The same phosphorylation that activates Dab1 also induces binding of a ubiquitin-conjugating enzyme (the Cullin-5-RING ligase complex, CRL5) to phosphorylated Dab1. CRL5 then catalyzes ubiquitin addition onto Dab1, and this stimulates Dab1 degradation by the proteasome. Dab1 activation thus sows the seeds for its own destruction. Critically, we found that Dab1 degradation is needed to shut down the Reelin response in the migrating cell. If this negative feedback loop is interrupted, neurons do not stop moving at the appropriate time and place, migration continues, and brain development is disrupted. These findings led to the concept that CRL5 provides a clock to time the duration of Src signaling in a given cell.
Learn more: Tyrosine kinase and ubiquitin signaling during brain development
Current Research
While we continue to investigate phosphorylation and ubiquitin signaling during development, our present major effort is to understand how CRL5 regulates tyrosine kinase signaling in other cell types. We found that inhibiting CRL5 in epithelial cells increases their migration and proliferation, and causes disorganized growth similar to cancer cells. These changes were abolished if Src was inhibited. This suggests that CRL5 normally inhibits Src signaling, presumably by stimulating the ubiquitylation and degradation of cell proteins that have been phosphorylated by Src. This leads to series of questions we are beginning to answer. For example: Which proteins stimulate cell migration and proliferation if they are not held in check by CRL5? CRL5 uses "substrate receptors", some of which bind phosphotyrosine residues and are known as SOCS proteins, to bind its substrates. So, which SOCS proteins (or other CRL5 substrate receptors) are involved in regulating cell growth and migration? And, is CRL5 providing a time clock to limit the duration of tyrosine kinase signaling under these conditions?
One of the CRL5 substrates we have identified is p130Cas (a.k.a. BCAR1), a known Src substrate that stimulates cell motility and proliferation. Interestingly, Cas also increases the resistance of breast cancer cells to anti-cancer drugs such as Tamoxifen. We found that phospho-Cas is targeted to CRL5 when the SOCS6 substrate receptor binds to a specific phosphosite in Cas. Detailed investigation showed that CRL5-SOCS6 targets phospho-Cas in focal adhesions at the leading edge of migrating cells, where the cells attach to the matrix. Our present studies are designed to better understand the dynamics of phosphorylation and ubiquitylation in focal adhesions, and to functionally characterize additional CRL5 phospho-substrates.
Learn more: Tyrosine kinase and ubiquitin signaling during cell migration and transformation
© 2025 Fred Hutchinson Cancer Center, a 501(c)(3) nonprofit organization.